FRC - A tool able to evaluate and rank de novo assemblies & assemblers.
Feature Response Curve
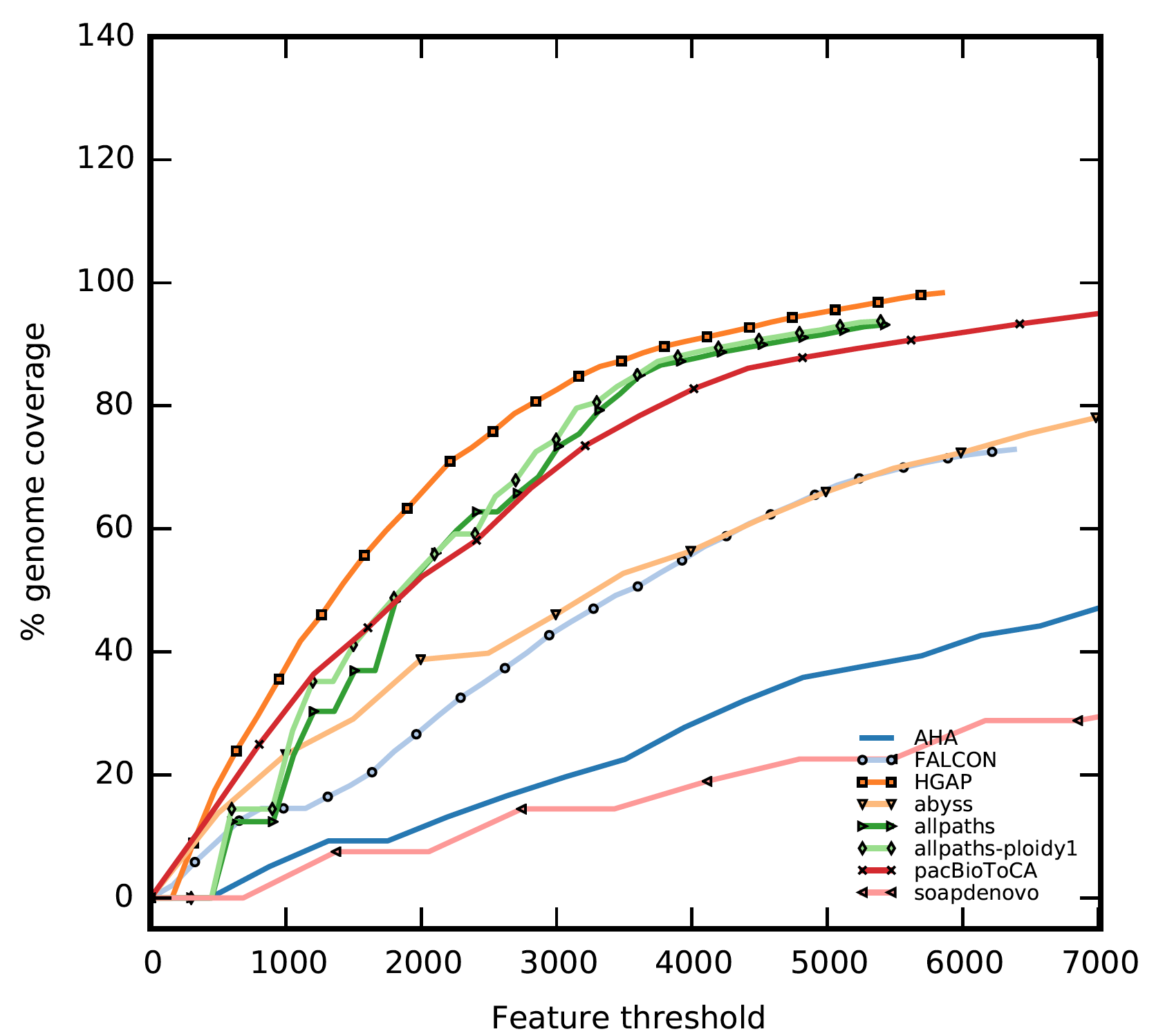
FRC uses anomalously mapped paired-end and mate-pair reads to identify suspicious areas, called features. Subsequently, features are tallied for each contig along with the estimated contig genomic-coverages. These points are ordered by decreasing contig size and plotted by accumulating the number of features. The resulting plot is in some aspects similar to a receiver operating characteristic (ROC), where the assembly with the steepest curve is likely to contain fewer mis-assemblies.
FRC has been successfully applied in several de novo assembly studies, including The Spruce Genome Project.
Installation
git clone https://github.com/vezzi/FRC_align.git
cd FRC_align
mkdir build
cd build
cmake ..
make
You will find the binaries in the main directory under bin. In case of problems the majority of the times there is a problem with the local installation of boost.
How to use FRC
Assemble your data (n PE libraries and m MP libraries) with your favourite tools. Let us call the assemblies A_tool1, A_tool2, etc.
- Align one PE library and one MP library against each of your assemblies (e.g., A_tool1)
- Use the same parameters
- PE library is mandatory, MP library is highly recommended
- sort and index the generated bam files by coordinate. We will call them A_tool1_PE_lib.bam and A_tool1_MP_lib.bam
- use PE library with largest read coverage (i.e., vertical coverage) and MP with largest spanning coverage (i.e., horizontal coverage)
- Run FRCurve for each assembly:
FRC --pe-sam A_tool1_PE_lib.bam --pe-min-insert MIN_PE_INS
--pe-max-insert MAX_PE_INS --mp-sam A_tool1_MP_lib.bam
--mp-min-insert MIN_MP_INS --mp-max-insert MAX_MP_INS
--genome-size ESTIMATED_GENOME_SIZE
--output OUTPUT_HEADER
where:
--pe-sam A_tool1_PE_lib.bam- sorted bam file obtained aligning PE library against assembly obtained with tool A
--pe-min-insert MIN_PE_INS- estimated min insert length
--pe-max-insert MAX_PE_INS- estimated max insert length
--mp-sam A_tool1_MP_lib.bam- sorted bam file obtained aligning MP library against assembly obtained with tool A
--mp-min-insert MIN_MP_INS- estimated min insert length
--mp-max-insert MAX_MP_INS- estimated max insert length
--genome-size ESTIMATED_GENOME_SIZE- estimated genome size
--output OUTPUT_HEADER- output header
Important: if --genome-size is not specified the assembly length is used to compute FRCurve. In order to be able to compare FRCurves
obtained with different tools (and hence producing slightly different assembly sizes) the same ESTIMATED_GENOME_SIZE
must be specified.
Output
OUTPUT_HEADER_Features.txt- human readable description of features: contig start end feature_type
OUTPUT_HEADER_FRC.txt- FRCurve computed with all the features (to be plotted)
OUTPUT_HEADER_FEATURE.txt- FRCurve for the corresponding feature
OUTPUT_HEADER_featureType.txt- For each featureType the specific FRCurve
Features.gff- Features description in GFF format (for visualization)
OUTPUT_HEADER_CEstats_PE.txt- CEvalues distribution (for CE_stats tuning)
OUTPUT_HEADER_CEstats_MP.txt- CEvalues distribution (for CE_stats tuning)
OUTPUT_assemblyTable.csv- Summary table containing general statistics on assembly coverage and insert size metrics
PE_lib_contigsTable.csv- Summary table containing per contig statistics (coverage, normal coverage, single coverage, etc) for PE library
MP_lib_contigsTable.csv- Summary table containing per contig statistics (coverage, normal coverage, single coverage, etc) for MP library
For more information and for advance use (i.e., CE-tuning) refer to the git-hub project page: https://github.com/vezzi/FRC_align
Contributors
Licence
GPL v3
See the code for FRC here: https://github.com/vezzi/FRC_align